统计之光--富集分析逻辑总结
GO富集分析
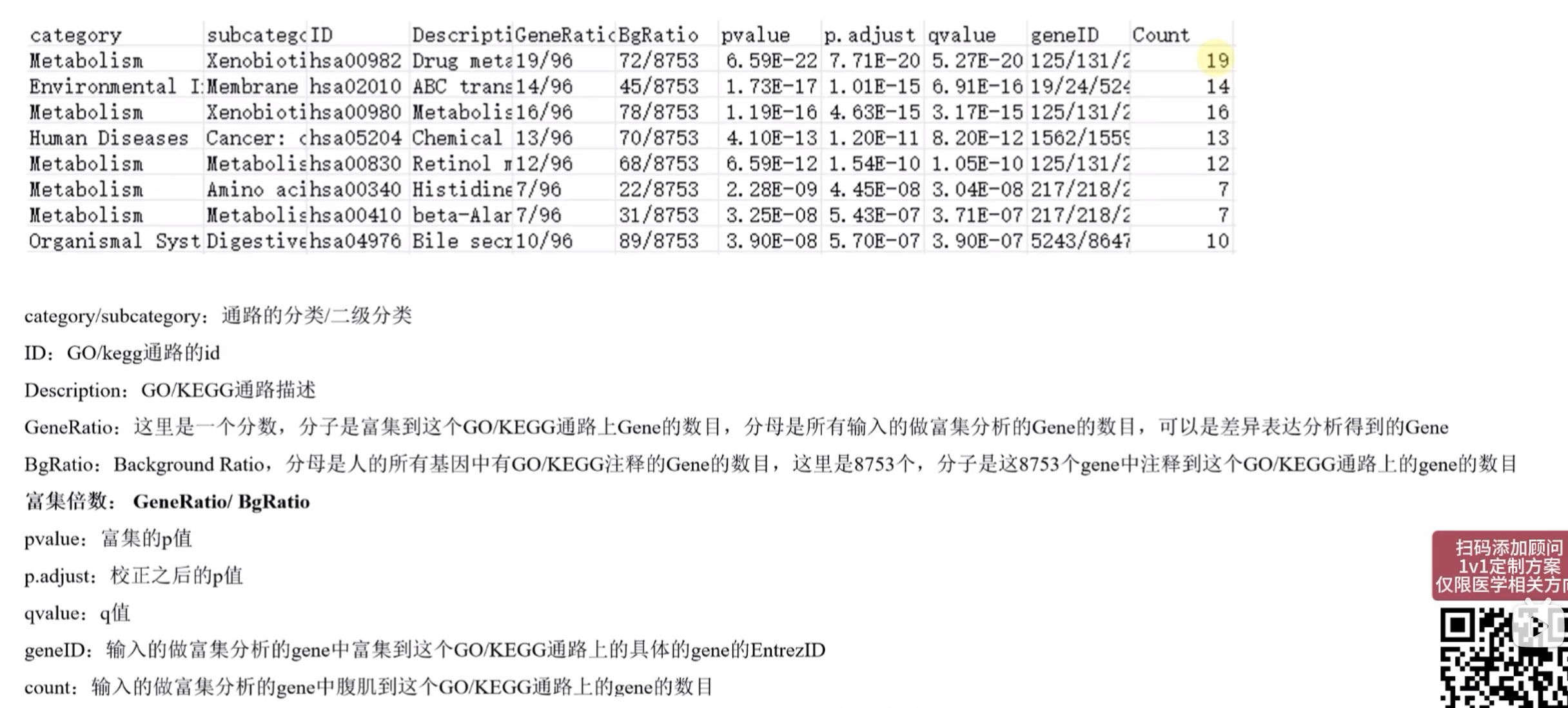
富集分析函数★
基因列表、物种基因数据库、基因名称类型、是否将基因ID转换为基因名字
enrich.go <- enrichGO(gene = special_gene, #待富集的基因列表
OrgDb = org.Hs.eg.db, #指定物种的基因数据库,示例物种是绵羊(sheep)
keyType = 'SYMBOL', #指定给定的基因名称类型,SYMBOL,ENSEMBL,ENTREZID三种类型
ont = 'ALL', #GO Ontology,可选 BP、MF、CC,也可以指定 ALL 同时计算 3 者
pAdjustMethod = 'BH', #指定用fdr作为 p 值校正方法
minGSSize = 10, # 富集的GO条目至少包含10个基因,可以修改
pvalueCutoff = 0.05, #指定 p 值阈值(<0.05)
qvalueCutoff = 0.2, #指定 q 值阈值(<0.2)
readable = FALSE) # 将GENE ID转为基因名字
go <- as.data.frame(enrich.go)
# 保存GO结果表
write.table(go, 'enrich.go.txt', sep = '\t', row.names = FALSE, quote = FALSE)
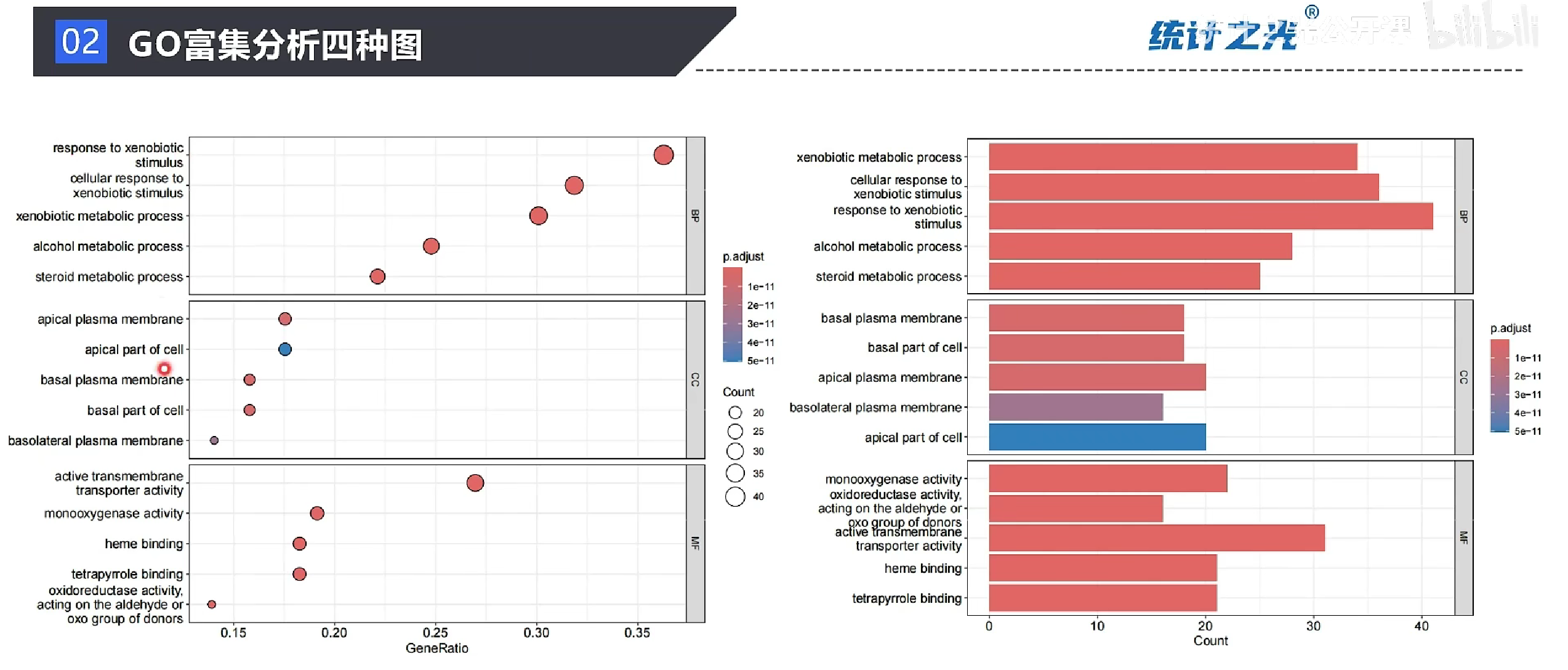
绘图
代码
library("ggplot2")
pdf(file='GO_ALL_DOT.pdf',width = 10)
dotplot(enrich.go,split="ONTOLOGY",showCategory=5)+facet_grid(ONTOLOGY~.,scale="free")
## 用ontology来分框,每个框里面显示5个条目(前五个)
dev.off()
pdf(file='GO_ALL_BAR.pdf',width = 10)
barplot(enrich.go,split="ONTOLOGY",showCategory=5)+facet_grid(ONTOLOGY~.,scale="free")
## 用ontology来分框,每个框里面显示5个条目
dev.off()
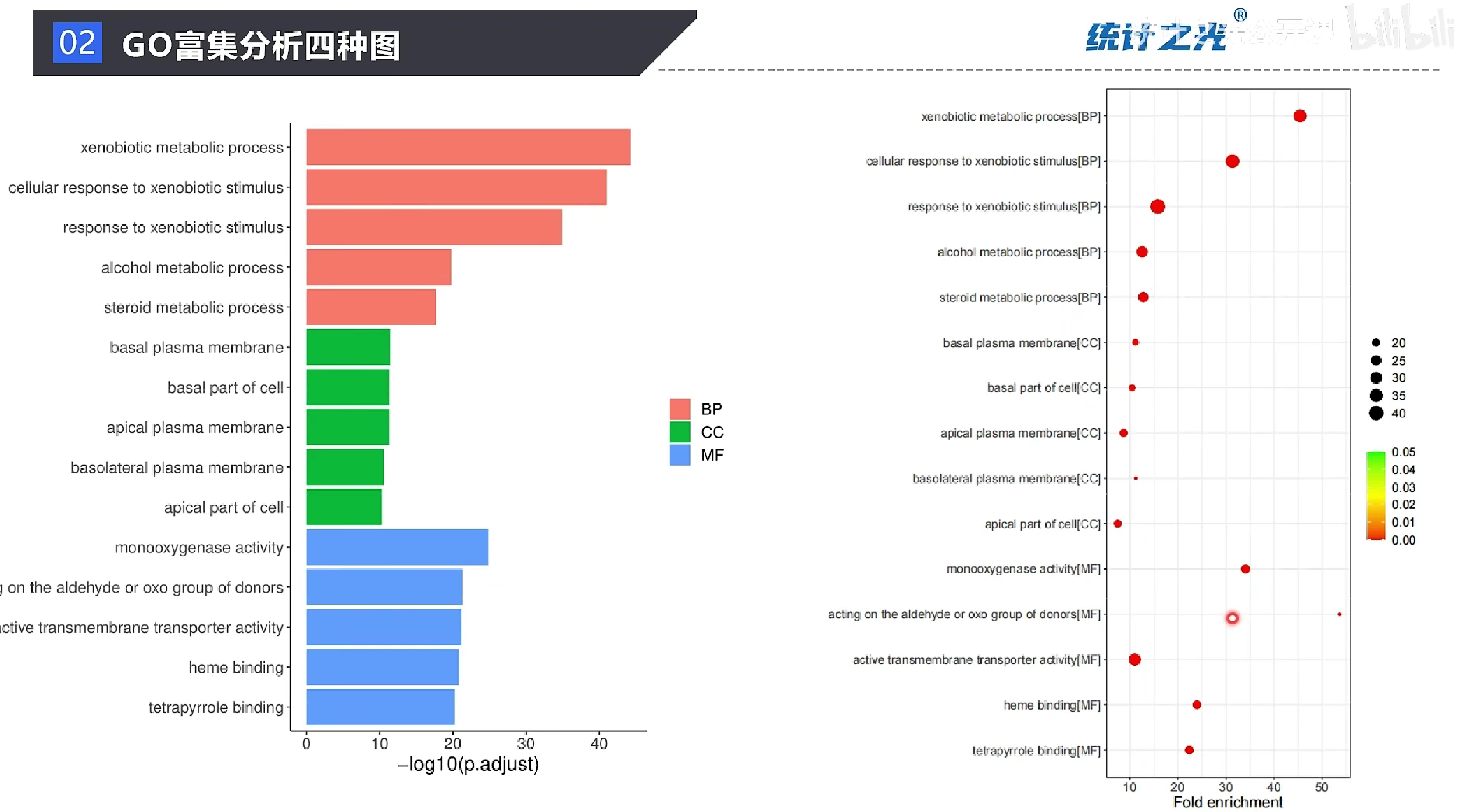
library(dplyr)
#挑选BP,MF,CC的前5个term,按校正后的p值,升序排列
top5 <- enrich.go %>% group_by(ONTOLOGY) %>% top_n(n=-5, wt=p.adjust)
top_5 <- as.data.frame(top5)
pdf("GO_barplot.pdf",width = 10)
#横轴是GO条目名字,纵轴是-log10(校正后的p值),根据BP,CC类别填充颜色
ggplot(top_5,aes(x=Description,y=-log10(p.adjust),fill=ONTOLOGY))+
geom_bar(stat="identity")+ #柱状图
coord_flip()+ # 交换横轴与纵轴位置
scale_x_discrete(limits=rev(top_5$Description))+ # 设置x轴上GO条目的顺序
theme_classic()+ # 采用经典主题
theme(
text=element_text(size=15), #所有的字体大小设置为20
axis.title.y=element_blank(), # y轴标签为空
axis.title.x=element_text(size=15), # x轴字体大小设为15
legend.title = element_blank() # 图标题为空
)
dev.off()
#定义将geneRatio和bgRatio转换成小数的函数
f2d <- function(ratio){
sapply(ratio,function(x) as.numeric(strsplit(x,"/")[[1]][1])/as.numeric(strsplit(x,"/")[[1]][2]))
}
#计算富集倍数,添加到go数据框最后一列
go_fe <- enrich.go %>% mutate(fe=f2d(GeneRatio)/f2d(BgRatio))
go_fe_df <- as.data.frame(go_fe)
#分别挑选BP,CC,MF的前5个term,按照校正后的p值,升序排列
top5_fe <- go_fe %>% group_by(ONTOLOGY) %>% top_n(n=-5, wt=p.adjust)
#每个GO条目后面加上对应的BP、CC或MF
top5_fe_term <- top5_fe %>% mutate(term=paste0(Description,"[",ONTOLOGY,"]"))
#转为数据框格式
top5_fe_term_df<-as.data.frame(top5_fe_term)
pdf("GO_ALL_DOT_inone.pdf",width = 10,height = 10)
ggplot(top5_fe_term_df,aes(x=term,y=fe,color=p.adjust,size=Count))+
geom_point()+ ##散点图
coord_flip()+ ## 交换x和y轴位置
scale_colour_gradientn(limits=c(0,0.05), #p值范围
colors = c("red","yellow","green"))+ #渐变颜色,p值越小,颜色越红
scale_x_discrete(limits=rev(top5_fe_term_df$term))+ #设置x轴上GO条目顺序
theme_bw()+ # 黑白主题
ylab("Fold enrichment")+ #y轴标题
theme(
text=element_text(size=15),
axis.title.y=element_blank(),
axis.title.x=element_text(size = 15),
legend.title = element_blank()
)
dev.off()
KEGG富集分析
分析函数
KEGG分析基因名称只能使用ENTREZID
此函数需联网查询
# 基因名称转换
entrezid_all = mapIds(x = org.Hs.eg.db, #id转换的比对基因组(背景基因)的物种,以人为例
keys = special_gene, #将输入的gene_name列进行数据转换
keytype = "SYMBOL", #输入数据的类型
column = "ENTREZID")#输出数据的类型
enrich.kegg <- enrichKEGG(gene = entrezid_all, # 这里需要输入的gene,ID只能未ENTREZID
organism = "hsa", #富集物种为人,物种缩写可参考:https://www.genome.jp/kegg/catalog/org_list.html
pAdjustMethod = "BH", #指定用fdr作为 p 值校正方法
pvalueCutoff = 0.05, #指定 p 值阈值(<0.05)
qvalueCutoff = 0.2) #指定 q 值阈值(<0.2)
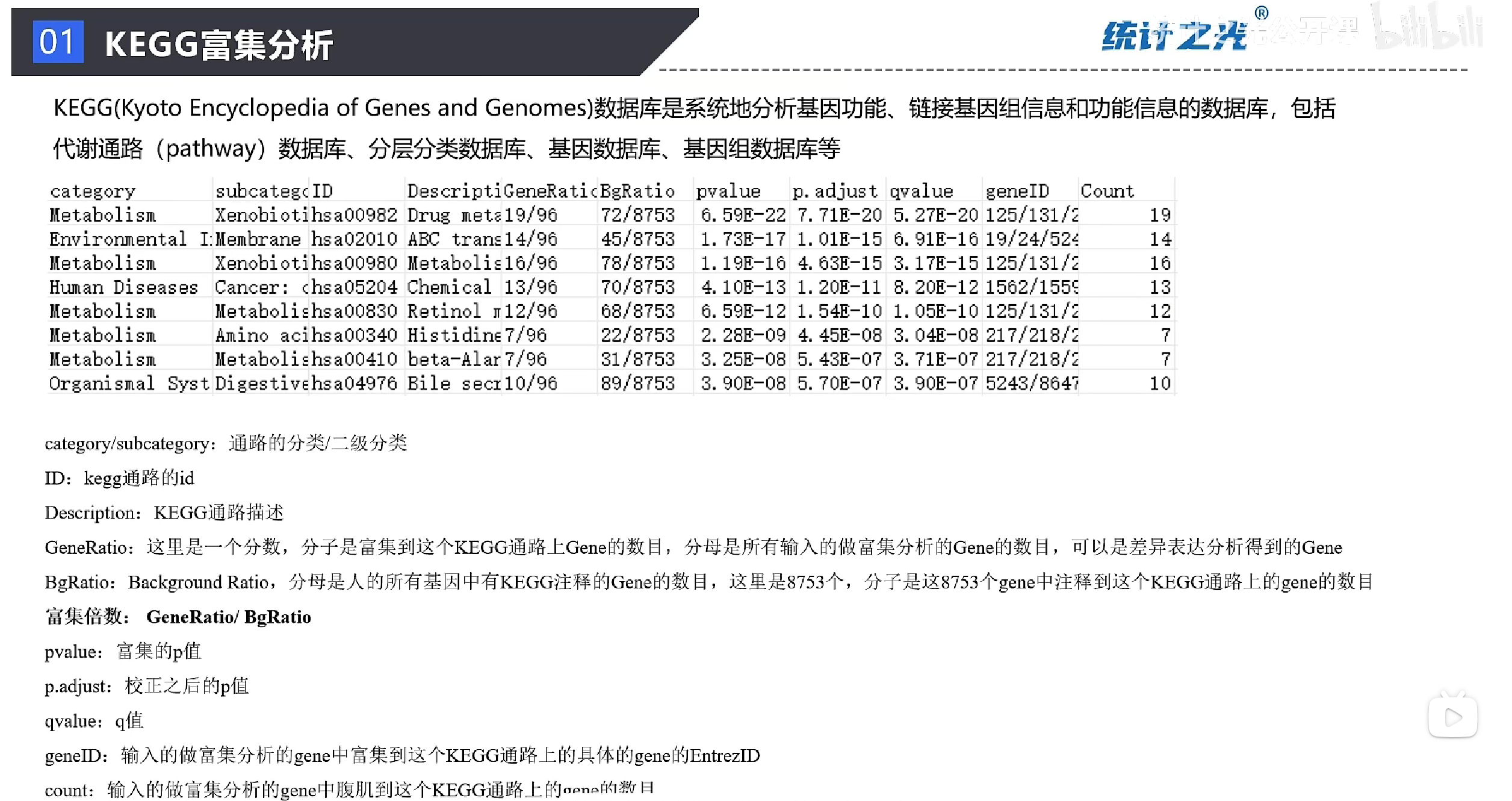
kegg_df <- as.data.frame(enrich.kegg)
write.csv(kegg_df,"kegg_enrich.csv",row.names = F)
展示结果
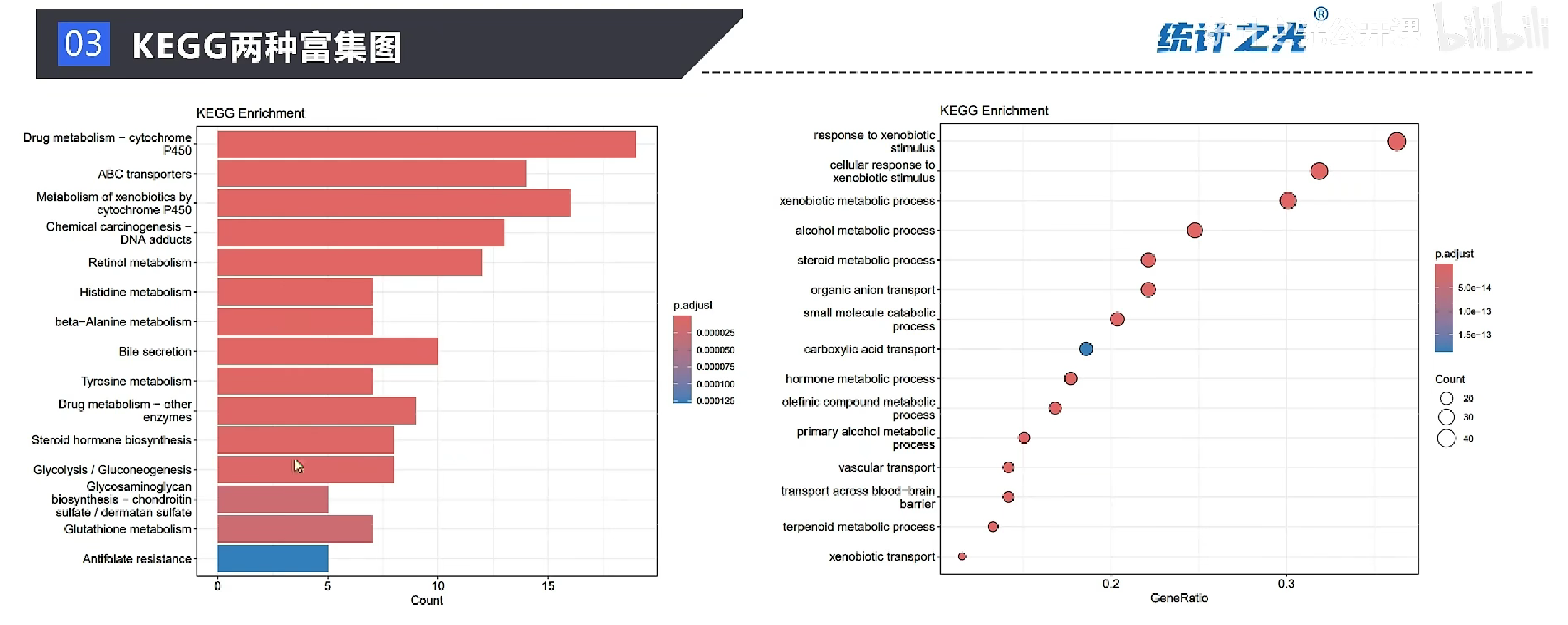
pdf(file = "KEGG_bar.pdf",width = 10)
barplot(enrich.kegg,showCategory = 15,title = "KEGG Enrichment")
# 展示前15个
dev.off()
pdf(file = "KEGG_dot.pdf",width=10)
dotplot(enrich.kegg,showCategory=15,title="KEGG Enrichment")
dev.off()
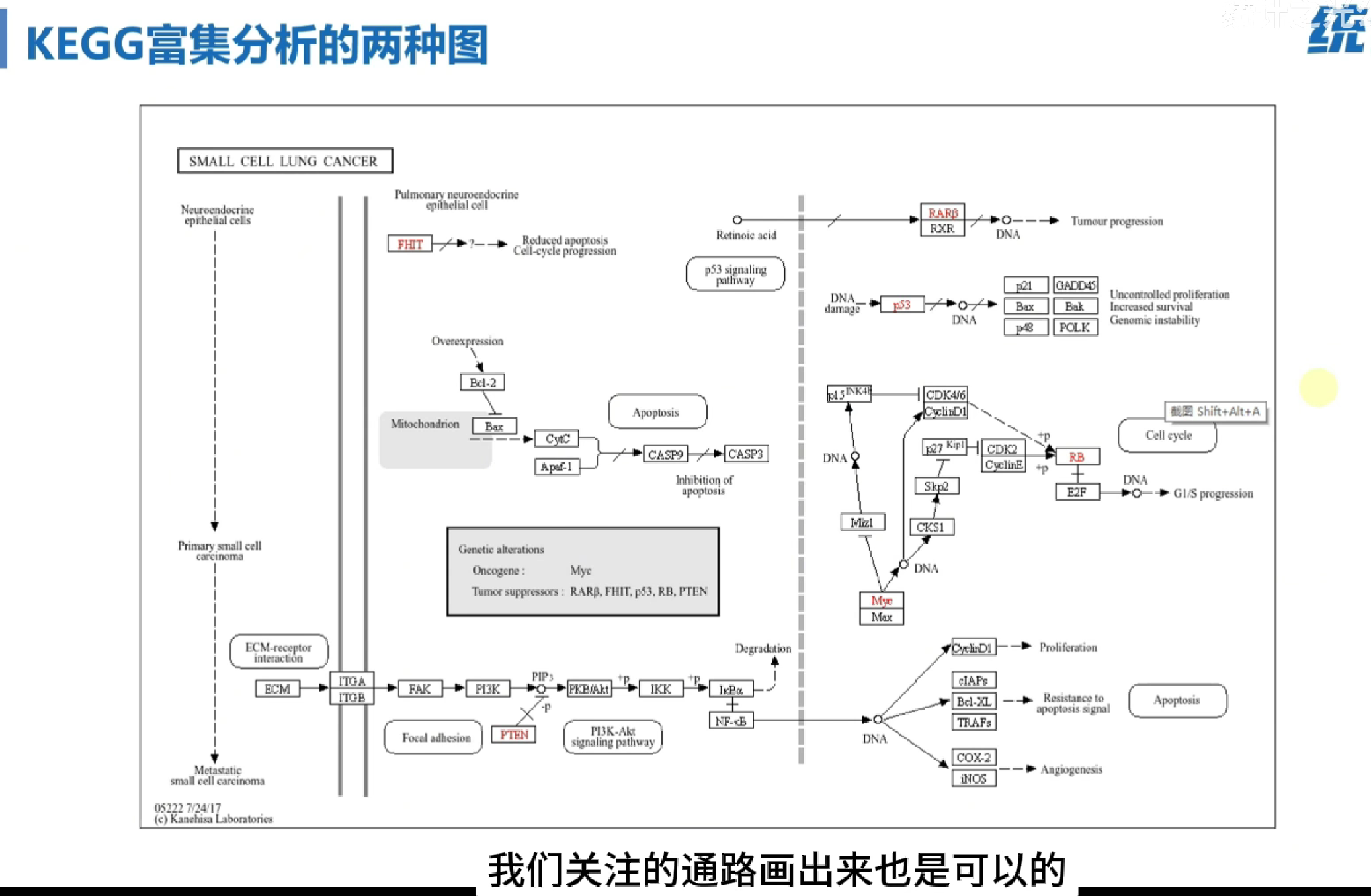
通路图
KEGG结果中的ID列——通路ID
绘图优点:标注出差异分析结果基因,及上下调水平
标注上下调水平需使用logFC
# BiocManager::install("pathview")
library(pathview)
# 2.3.1 简单版
## 以hsa00982通路举例,此处标记了所筛选基因
## 将所筛选基因列表、通路ID作为输入项
hsa00982 <- pathview(gene.data = special_gene, #基因列表
pathway.id="hsa00982", # 通路名
species ="hsa", #物种
gene.idtype= "SYMBOL", #基因id或名称类型,SYMBOL,ENSEMBL,ENTREZID 三种类型
kegg.native=TRUE) #TRUE:输出png图片,FALSE:输出pdf文件
# 2.3.2 标注上下调水平版
## 此处还标记了所筛选基因的上下调水平
## 将行名为基因名,列名为logFC的数据、通路ID作为输入项
deg_kegg1 <- resSig[special_gene,]
deg_kegg2 <- deg_kegg1$log2FoldChange
view(deg_kegg2)
names(deg_kegg2) <- special_gene
hsa00982 <- pathview(gene.data = deg_kegg2,
pathway.id="hsa00982",
species ="hsa",
gene.idtype= "SYMBOL",
kegg.native=TRUE,
limit=list(gene=max(abs(deg_kegg2)),cpd=1)
)
GO富集分析介绍

KEGG富集分析介绍